Overview
In this exercise, we’ll modify the original Hardy-Weinberg model to include the effects of inbreeding, which is covered in Chapter 13 of your text (we’ll get to this after break).
Remember that in the original HW model, we combined gametes at random, according to their probabilities. But we know that mating is not always random! To break that assumption, we’ll need to change our flows to incorporate non-random mating. We could model a variety of forms of non-random mating but the most common one that we encounter in nature is inbreeding so that’s what we’ll work on here.
Recall that we are starting with a single locus and 2 alleles, continuing to model non- overlapping generations (adult die immediately after producing gametes). With random mating, every individual contributes gametes to the gene pool and then those are combined at random (following HW expected values) to form new zygotes. In contrast, inbreeding means that individuals will mate with relatives more often than expected by chance. Which is to say, they will more often than expected from HW proportions! So we’ll clearly need to alter our flows (where gametes are paired into zygotes) to adjust for inbreeding.
To keep things simple, we’ll consider selfing so that A1A1 individuals are more likely to mate with themselves, thus yielding more A1A1 zygotes than otherwise expected. And similarly for the other two genotypes. Note that this tendency to mate with oneself will not change allele frequencies, it will just reshuffle the existing alleles, thus changing genotype frequencies. Since allele frequencies won’t change but genotype frequencies will, we can estimate the inbreeding coefficient (F) to describe how inbred our population is compared to HW expectations for the given allele frequencies.
In natural populations, selfing is pretty normal, especially in plants where hermaphroditism is common. However, populations often display a mixed-mating system, meaning that individuals produce some offspring via inbreeding (such as selfing) and then some of their offspring result from outcrossing with unrelated mates. For example, in the annual plant jewelweed (Impatiens spp.), plants produce two types of flowers, cleistogamous (which pollinate themselves before opening) and chasmogamous (outcrossing flowers). These flower types even grow at different places on the plant - small cleistogamous flowers are close to the stem, pollinate themselves without even opening up, and drop their seeds at the base of the plant. Meanwhile, large, colorful chasmogamous (outcrossing) flowers are at the ends of branches, have only pollen or stigma receptive at a time, and produce seeds that burst explosively from their pods when ripe. An individual plant can thus adjust how much it inbreeds versus outcrosses based on how many of each type of flower it produces!
We’re going to incorporate this mixed-mating strategy into our model since the rate of inbreeding is likely to have consequences for how much genotypic diversity is depleted. You will also see that if we assume that the population does a mixture of inbreeding and outcrossing, then we will expect to reach an equilibrium that does still include heterozygotes.
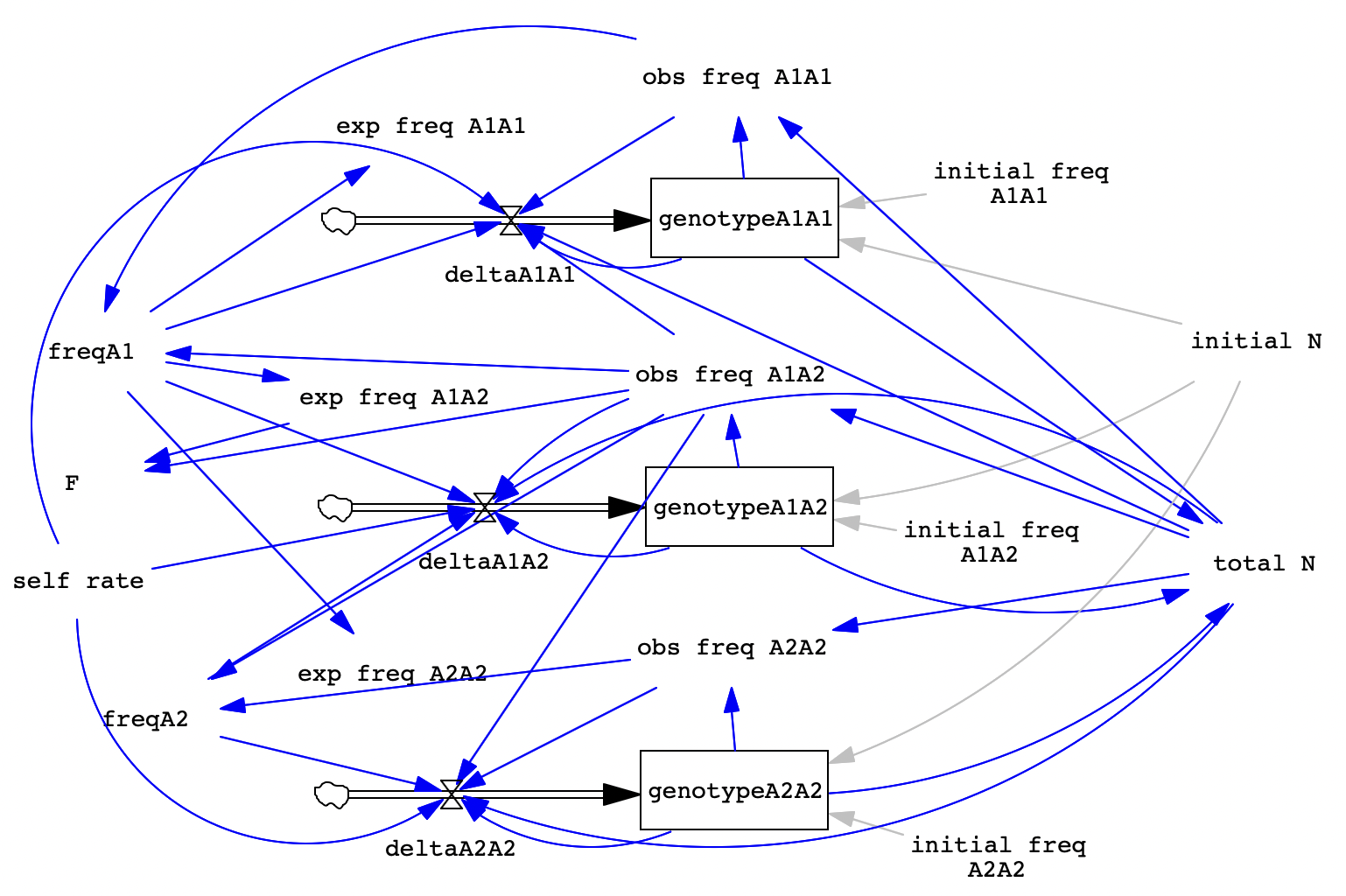
Here’s an image of the completed model for your reference.